Recently, we exceeded 40,000 gene lists in our database. At some point in the (not-so-near) future, we'll upgrade the very basic appearance of our site. However, the majority of our labor has been, and will be, focused on the underlying database. More lists means more opportunities for the user to find studies that strongly intersect with his/her own lists. It means more chances to find genes that are significantly co-expressed with the user's own genes of interest. Etc.
40,000 lists means approximately 800,000,000 study/study intersections, each with an associated P-value. When we break the 50,000 mark, we'll have about 1,250,000,000 intersections. Here, a 25% increase in database size means greater than 50% more P-values. For biological truthseekers and hypothesis-generators, database size should be critical, not a pretty interface.
We'd also point out that, with few exceptions, our database is not littered with recycled GO lists and the like. Most of our lists will not be found elsewhere. Sometimes I get the feeling that a large portion of gene enrichment tools are generated by folks whose primary interest is in programming and computer science, not biology. The database content is thus an annoyance that must be dealt with. The easy solution to this annoyance is to grab existing GO lists and manufacture some new, tricky algorithms that make the tool worthy of an NAR paper.
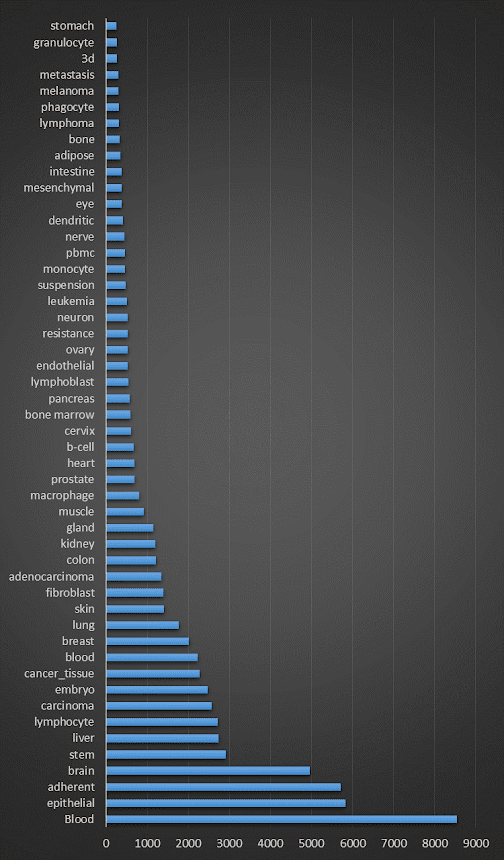
So, what's in the February 2022 incarnation of the database? First, let's look at the species breakdown:


In the case of the terms "PTM", "methylation", "antigen", "chip", and "epitranscriptome", we list the genes associated with these events. Some would dispute the inclusion of such studies within our database. A hypermethylation event, of course, has a very specific location. A nearby location on the same gene could be hypomethylated, meaning that this gene could be found in both hyper- and hypo-methylation lists from a single study. We justify this approach with the simple observation that two hypermethylation lists from different studies may overlap very significantly (try it: find a list of hypermethylated genes in a particular cancer type and enter it into our Fisher tool...don't bother selecting any options under the "molecule" filter).

